Secondary Schizophrenia (86 page)
Read Secondary Schizophrenia Online
Authors: Perminder S. Sachdev

15q [15]
, mutations of which cause absent or low
tation appears to be psychosis
[19, 29,
32, 33, 34].
Rates
levels of HEX-A
[16]
and produce a predominantly
of psychosis in late-onset TSD patients range from 30%
neurological syndrome. Mutations of the
β
–subunit
[33]
to 50%
[32].
In a review of all published cases
gene on chromosome 5 cause low levels of both HEX-before 1998, MacQueen
[27]
estimated the conserva-A and HEX-B
[17]
and cause GM2-gangliosidosis
tive prevalence at 33% of late-onset patients. The form
type II (Sandhoff Disease), where the viscera as well
of this psychosis has been described as “hebephrenic”
as the CNS is affected
[18].
Mutations to different
with disorganization, auditory and visual hallucina-regions of the
α
–subunit gene result in variably sta-tions, but also clouding of consciousness and cognitive
ble transcripts and thus variable resultant enzyme lev-impairment. Cases have been described where the pre-els, which may correspond to the variable onset of
senting feature was catatonia
[30, 35, 36].
the disease
[14, 19].
In most adult-onset patients the
Treatment of psychosis in TSD appears problem-mutations G269S or W474C are present
[19].
HEX-atic, with often only partial response to neurolep-A deficiency in lysosomes impairs the catabolism
tics and mood stabilizers such as lithium
[27,
30,
of gangliosides from the neuronal cell membrane,
36, 37]
. Importantly, patients with late-onset TSD are
resulting in accumulation of lysosomal gangliosides.
exquisitely sensitive to the motor side effects of many
This leads to secondary neuronal changes, particu-neuroleptic medications
[30, 36].
For severe psychotic
larly axon hillock outgrowth to form “meganeurites”
or affective illness, electroconvulsive therapy (ECT)
with ectopic dendritogenesis
[20]
and focal axonal
appears to be a safe and effective treatment to offer this
216
enlargements known as “axonal spheroids”
[21],
both
patient group
[35, 37].
Chapter 16 – Storage disorders and psychosis
Niemann-Pick Disease type C
Niemann-Pick Disease type C (NPC) is an autosomal
recessive neurovisceral disorder of lipid storage, with
a frequency of 1 in 100,000 live births
[38].
It is
characterized by variable degrees of cognitive decline,
behavioral disturbance, and neurological impairment,
predominantly ataxia, and vertical supranuclear
opthalmoplegia
[39].
It is biochemically and pheno-typically distinct from Niemann-Pick Disease types
A and B, which result from a deficiency of lysosomal
sphingomyelinase
[40, 41].
Genetic analysis reveals
two distinct genetic foci, with 95% of the disease
caused by aberrations in the NPC1 gene on 18q11–12
[42],
coding for the lysosomal NPC1 protein
[43].
The
less common NPC2 variant is caused by mutations
in the NPC2 gene, mapping to chromosome 14q24.3
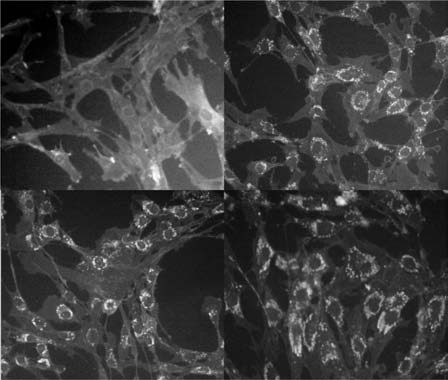
Figure 16.1
Filipin staining of cultured fibroblasts in Niemann-Pick
[44]
and whose product resides in the Golgi apparatus
Disease type C. Top left shows normal cells with minimal staining;
top right and bottom left and right show staining of perinuclear
and late endosomes. These proteins are involved in
cholesterol in three NPC-sufferers who presented with psychosis in
cyclical movement of sterols within cells
[45, 46, 47],
adulthood and are described in Walterfang et al., 2006 [86].
performing cholesterol trafficking and homeostatic
functions
[48, 49].
Mutation and dysfunction of NPC1 and NPC2
The diagnosis of NPC can be confirmed by demon-appear to result in late endosomal accumulation of
strating a low esterification rate of exogenous choles-cholesterol, some glycolipids, and selected ganglio-terol in cultured skin fibroblasts
(Figure 16.1),
or by
sides
[50, 51]
leading to Alzheimer-like neurofibrillary
testing for lysosomal accumulation of free cholesterol
tangles (NFTs), neuronal degeneration, neuroaxonal
by filipin staining
[67].
The “classical” biochemical
dystrophy, and demyelination
[47, 52, 53, 54].
This
phenotype shows markedly reduced esterification and
intracellular cholesterol “traffic jam” impairs the trans-greater than 70% to 80% of cells staining positive for
port of endogenously synthesized cholesterol to dis-filipin, whereas the “variant” phenotype shows near-tal axons, where it is required for membrane mainte-normal esterification rates and lower filipin-positive
nance
[55]
and response to axonal injury
[56].
Axonal
cell counts while still demonstrating clinical symptoms
structures are therefore particularly vulnerable and
[67].
are affected early with axonal spheroid formation,
NPC may present in infancy, adolescence, or adult-hypomyelination, and eventual demyelination
[57].
As
hood
[68]
with a clinically variable picture, although
a result, white-matter tracts are severely affected
[51,
its core features include dementia, dysarthria, ataxia,
58, 59]
, with the corpus callosum showing the most
vertical supranuclear opthalmoplegia, and hep-striking axonal loss
[60].
atosplenomegaly. It may also commonly present
The neuronal cells most vulnerable to NFT accu-with dystonia and choreoathetosis
[68, 69].
Seizures,
mulation are the Purkinje cells of the cerebellum,
dysphagia, and pyramidal signs may appear with
basal ganglia, and thalamus followed by neurons in
disease progression. The range of NPC1 and NPC2
hippocampal and cortical regions
[59,
61, 62, 63].
mutations results in marked heterogeneity of clinical
Affected neurons often show ectopic dendritogene-presentations
[70].
sis with stunted dendrites and greatly reduced den-Structural imaging in NPC commonly shows
dritic arborization
[64].
Altered phosphorylation of
diffuse cerebral and/or cerebellar atrophy
[68, 71,
the microtubule-associated protein
MAP2
results in
72, 73, 74]
or callosal pathology
[58,
75]
(Figure
dendritic microtubule depolymerization
[65],
and a
16.2). Occasionally, white matter hyperintensities may
reduced availability of arborization-promoting neu-present
[68, 74, 75, 76, 77],
which may radiologically
rosteroids secondary to cholesterol unavailability
mimic multiple sclerosis
[75].
Single photon emis-
217
sion computed tomography (SPECT) and positron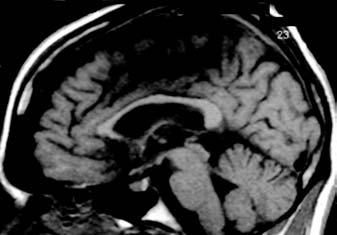
Organic Syndromes of Schizophrenia – Section 3
(B)
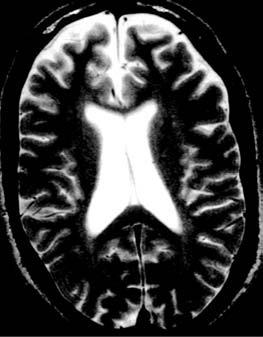
Figure 16.2
Magnetic resonance
imaging scan in Niemann-Pick Disease
type C in a 23-year-old patient with
adult-onset disease and a 10-year history
of schizophrenia-like psychosis. Left, a
(A)
sagittal T1-weighted image showing
callosal thinning; right, a T2-weighted axial
section showing frontal atrophy.
emission tomography (PET)
[78]
imaging may show
disorder that responded to anticonvulsant medication
hypoperfusion in frontal regions
[77, 79, 80],
whereas
[90].
magnetic resonance spectroscopy (MRS) in psychotic
and nonpsychotic patients shows reductions in N-acetyl-aspartate:creatine ratios suggestive of pathol-
Cerebrotendinous xanthomatosis
ogy in the frontal and parietal cortices and basal
Cerebrotendinous xanthomatosis (CTX) is a rare auto-ganglia
[72, 74],
where changes appear to correlate
somal recessive disorder caused by mutations of the
with clinical dysfunction
[74].
Some of these features
serol-27-hydroxylase gene on 2q35
[91],
which results
overlap with those found in schizophrenia, includin increased tissue cholestanol and defective bile
ing hypofrontality
[81],
striatal pathology
[82],
and
acid synthesis
[92]
. Accumulation of cholestanol in
white-matter changes
[83].
Electro-encephalography
white and grey matter leads to neuroaxonal dystro-
(EEG) may demonstrate diffuse slowing
[68, 73, 80,
phy and possibly accelerated apoptosis
[93].
Individ-
84].
Neuropsychological testing in adolescent/adult-uals with CTX develop xanthomas, cataracts, men-onset cases often reveals a steady decline in function
tal retardation, or dementia, and varying movement
throughout adulthood with significant deficits in exec-disorders. Global reduction in grey-and white-mat-utive function and memory
[71, 72, 80, 84].
ter volume, reduced white-matter intensity, and cal-Psychosis is not an uncommon sign later in the
losal atrophy are noted on structural imaging
[94, 95],
presentation of adolescent or adult-onset NPC, but in
which may – like NPC – mimic the demyelination
25%–40% of adult-onset cases it may present alongside
of multiple sclerosis
[96].
MRS findings suggest that
motor symptoms and cognitive impairment as an ini-axonal metabolic dysfunction rather than demyelinatial manifestation
[85, 86].
When psychosis has been
tion is responsible for the diffuse white-matter findings
reported, features have included persecutory delu-
sions, auditory hallucinations, and ideas of reference,
Two case series of CTX patients with psychias well as behavioral disorganization
[71, 80, 84, 85,