Wallach's Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis (297 page)
Authors: Mary A. Williamson Mt(ascp) Phd,L. Michael Snyder Md

Serum erythropoietin is elevated.

HEMOGLOBINOPATHIES
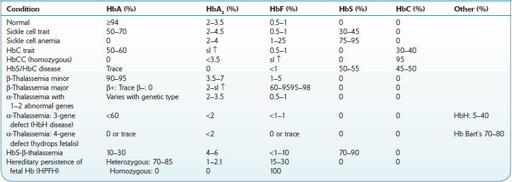
Hemoglobinopathies constitute the most common inherited disorders in humans as a result of selective pressure of endemic falciparum malaria. Human hemoglobins (Hb) are proteins containing a heme moiety and two pairs of globin genes. Normal adult Hb is composed of two alpha and two β-chains, which together add up to 97% of total Hb in red cells (RBC). The balance globins are composed of Hb A2 (approximately 2.5%) and fetal Hg (HbF) usually 0.8–2%. More than 1,000 mutations involving the globin genes have been described; they result from amino acid substitution or from abnormalities of synthesis. The majority of these variants do not cause clinical or hematologic problems. Several variants, such as sickle cell disease and β-thalassemias (described below), are protective and asymptomatic in the heterozygous; however, they result in severe morbidity in the homozygous. Initial screening and definitive diagnosis for Hb variants are described in Chapter
16
. Table
9-1
describes the most common hemoglobinopathies encountered in North America: sickle cell syndromes, HbC disease, and β- and α-thalassemias. Genetic analysis may be necessary for uncommon or unknown variants. In North America, it is done in a few specialized laboratories.
TABLE 9–1. Hemoglobinopathies
SICKLE CELL ANEMIA
Definition
The term sickle cell disease (SCD) describes all the conditions associated with sickling of RBC. SCD encompasses a group of conditions with autosomal inheritance of abnormal Hb β chain resulting from a substitution of valine for glutamic acid in the beta globin chain. This substitution results in polymerization of poorly soluble deoxy-HbS, leading to a marked decrease in red cells’ deformability and irreversible distortion of red cells into the sickle cell shape, with their removal by the spleen (prior to the occurrence of autosplenectomy) and macrophages. SCD is encountered mostly in populations of African or Arab ancestry, as well as in some Indian groups.
Aplastic crises
are self-limited episodes of erythroid aplasia lasting 5–10 days. They are due to infections (most commonly parvovirus B19) and may require emergency transfusions.