Wallach's Interpretation of Diagnostic Tests: Pathways to Arriving at a Clinical Diagnosis (217 page)
Authors: Mary A. Williamson Mt(ascp) Phd,L. Michael Snyder Md

The presence of symptoms or signs suggestive of hormonal activity warrants further evaluation with appropriate biochemical screening tests (Figure
6-7
; Table
6-4
).
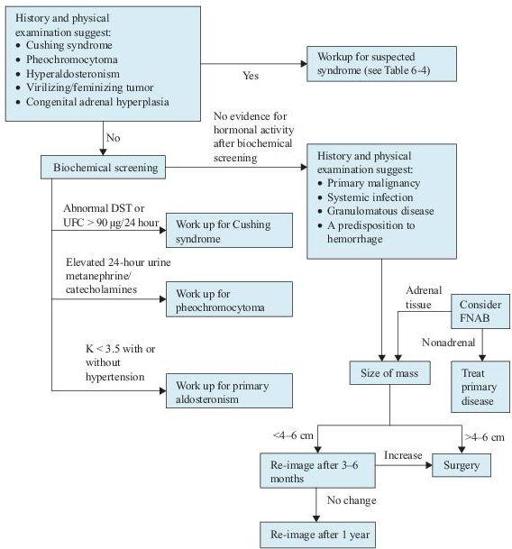
Figure 6–7
Algorithm for the diagnosis of adrenal masses. Consider both clinical presentation and appearance on imaging in deciding cutoff size. DST, dexamethasone suppression test; FNAB, fine needle aspiration biopsy; UFC, urinary free cortisol.
TABLE 6–4. Clinical Presentations of Hormonal Hypersecretion and Recommended Screening Tests
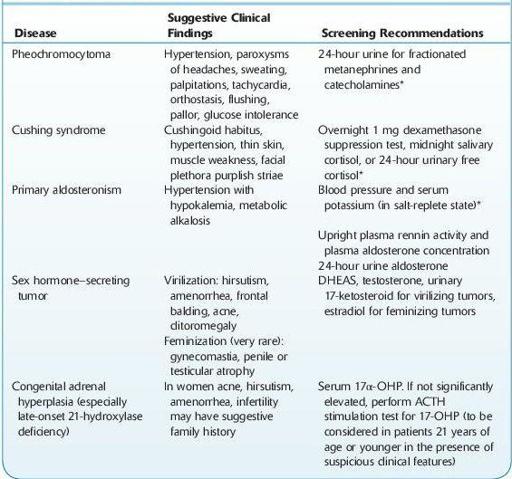
*Screen in all patients with an incidental adrenal mass.
ACTH, adrenocorticotropic hormone; DHEAS, dehydroepiandrosterone sulfate; 17α-OHP, 17-hydroxyprogesterone.
Laboratory Findings
1. The goal of evaluation is to determine which masses are functional and which ones have the likelihood of being malignant. Benign tumors without hormonal activity need only to be followed, whereas most hormonally active and primary malignant tumors need to be removed. Appropriate biochemical screening tests are listed in the table and algorithm. In patients with no apparent signs or symptoms, a basic biochemical screening is necessary because up to 11% of the cases will have unsuspected abnormal adrenal function.
2. CT and MRI are helpful in determining the likelihood of the adrenal masses being malignant. Size is the most important predictor. Masses >4–6 cm are recommended for surgical removal. Close follow-up is recommended for smaller masses for any change in size.
3. FNA biopsy can differentiate adrenal from nonadrenal masses, but not benign from malignant adrenal tissue. Therefore, it is most useful for evaluation of metastatic disease in patients with a known or suspected cancer outside the adrenal gland.
Suggested Readings
Khan F, Sachs H, Pechet L, et al.
Guide to Diagnostic Testing
. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al.
Williams Textbook of Endocrinology
, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc.; 2008.
Lacroix A. Clinical presentation and evaluation of adrenocortical tumors. In: Rose B, (ed).
UpToDate
, Waltham, MA: UpToDate, Inc.; 2009.
Young WF Jr, Kaplan NM. The adrenal incidentaloma. In: Rose B, (ed).
UpToDate
, Waltham, MA: UpToDate, Inc.; 2009.
PHEOCHROMOCYTOMA
Pheochromocytoma refers to catecholamine-secreting tumors arising from the chromaffin cells of the adrenal medulla or the sympathetic ganglia (extra-adrenal).
Pheochromocytomas are rare neoplasms with an annual incidence of 2–8 cases per 1,000,000 people. This entity constitutes <0.2% of patients with hypertension. These tumors are curable when diagnosed and treated properly, but they are potentially fatal if missed.
The 10% rules refers to the following: 10% pheochromocytomas are extra-adrenal, 10% are seen in children, 10% are bilateral, 10% represents recurrence, 10% are malignant, and 10% are familial. Familial syndromes include
A. Familial pheochromocytomas
B. Multiple endocrine neoplasia (MEN) type 2
MEN type 2A: pheochromocytoma, medullary carcinoma of the thyroid, and hyperparathyroidism
C. Neurofibromatosis 1 (NF1). The main features of NF1 are neurofibromas and dermal café-au-lait spots. NF1 has been associated with a variety of endocrine neoplasms including pheochromocytoma, somatostatin-producing carcinoid tumors of duodenal wall, medullary carcinoma of thyroid, and hypothalamic or optic nerve tumors.
D. von Hippel-Lindau disease (VHL). It is an autosomal dominant neoplastic syndrome characterized by hemangioblastomas of central nervous system, retinal angiomas, renal cell carcinomas, visceral cysts, pheochromocytoma, and islet cell tumors.
The classic triad of symptoms includes episodic headache, sweating, and tachycardia. However, not all patients have the three classic symptoms, and patients with essential hypertension may have the same symptoms. Therefore, pheochromocytoma should be suspected in patients who have one or more of the following:
1. Sustained or paroxysmal hypertension
2. Generalized sweating, palpitations, headache, tremor, and panic attack–type symptoms
3. Familial syndrome of MEN2, NF1, or VHL
4. Family history of pheochromocytoma
5. An adrenal mass incidentally discovered by imaging studies